
美国已经形成了较为完整成熟的临床试验注册和结果信息制度,任何人可以在ClinicalTrials.gov网站检索到可靠的临床试验数据,该制度也进一步推动了美国临床医学在全球的影响力。
本文将针对向ClinicalTrials.gov网站提交临床试验注册和结果数据共享政策作简要综述。
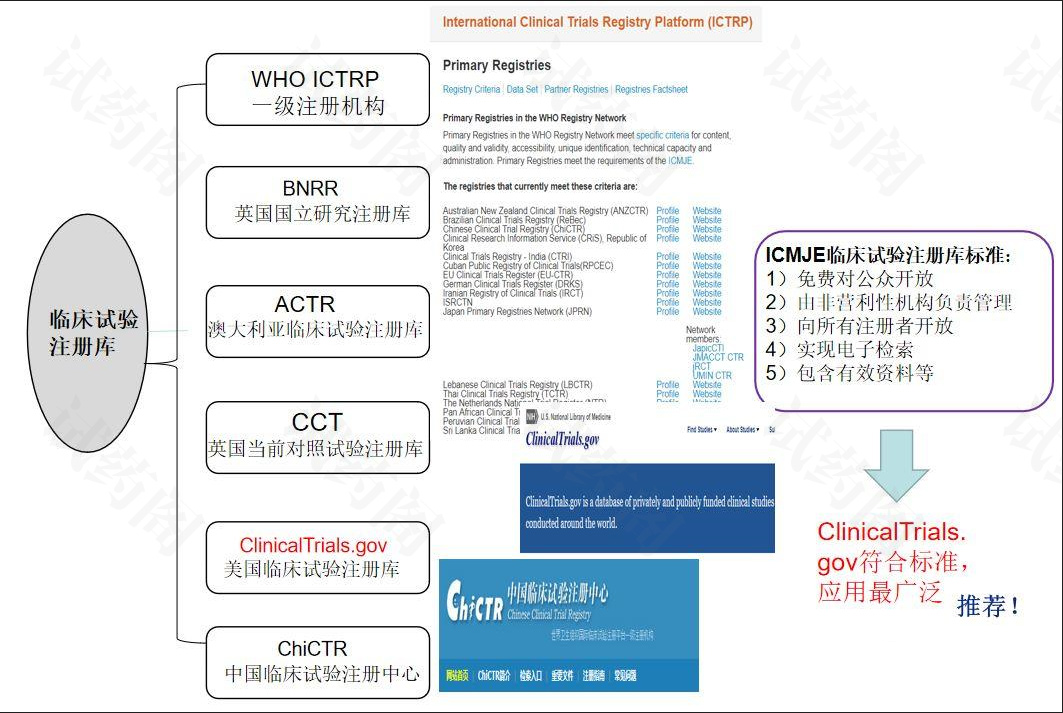
1 美国临床试验注册和结果提交信息制度
1997年美国《食品和药品管理现代化法》(Food and Drug Administration Modernization Act (FDAMA) of 1997)规定“应建立、维护和运行一个用于针对严重或危及生命的疾病和药物临床试验信息数据库,该数据库的活动应与美国卫生与公共服务部(Department of Health and Human Services, (HHS))活动相协调并在可行的范围内协调与其类似的其他数据库”。2
为响应该规定,美国国立卫生研究院国家医学图书馆(National Library of Medicine, (NLM))开发了名为ClinicalTrials.gov的数据库。该网站于2000年2月公开使用。
《2007年食品和药品管理局修正法案》(Food and Drug Administration Amendments Act of 2007 (FDAAA))第八章(以下简称FDAAA 801)修改《公共卫生服务法》(Public Health Service Act)第402(j)节3,增加“扩大临床试验登记数据库”内容,要求FDA监管的符合特定要求的药品、生物制品和器械产品的临床试验应当在ClinicalTrials.gov网站注册并报告结果信息摘要。自此美国建立了以网上注册和公开研究结果为核心的临床试验注册和结果提交制度4。
自ClinicalTrials.gov建立以来,科学界、政府机关和社会组织均采取相应的措施,以提高临床试验信息的可及性和透明度。由临床试验发起人以及主要研究者提交和维护网站数据库中的临床试验研究数据,并向医疗卫生人员及社会公众提供临床试验查询服务的模式也被各国争相模仿5。经过多年补充完善,ClinicalTrials.gov已经成为最具国际影响力的开放性临床试验数据库之一。6
2 《临床试验注册和结果信息提交的最终规则》主要内容
2016年9月,HHS发布《临床试验注册和结果信息提交的最终规则》(以下简称《最终规则》),阐明并扩展了根据FDAAA801向ClinicalTrials.gov 提交某些试验的注册和结果信息的监管要求和程序,旨在向申办者、调查人员和公众明确哪些试验必须报告,何时必须报告,以及是否已达到要求。7该规则于2017年1月18日生效。
《最终规则》规定,适用的临床试验(Applicable Clinical Trial, (ACT))的责任方必须在ClinicalTrials.gov网站提交该临床试验的信息,向社会共享。
2.1. 适用的临床试验(ACT)
受最终规则强制约束的ACT须满足两个条件。
第一,ACT是受美国FDA监管的药物、生物制品或者器械产品。8其中,适用器械临床试验包括《联邦食品、药品和化妆品法》(21 U.S.C. §§ 301 ET SEQ.)第510(k)、515或520(m)条规定的针对人类受试者对照的器械产品临床研究9、《联邦食品、药品和化妆品法》第522条规定的对器械产品进行儿科上市后监督(A pediatric postmarket surveillance of a device product)10、《联邦法规法典》(code of federal regulation)第三部分规定的组合产品的临床试验11。适用药品的临床试验指《联邦食品、药品和化妆品法》第505条规定的药物产品12或《公共卫生服务法》第351条规定的除第一期临床研究外的生物制品受控临床调查13。《最终规则》更细化规定,ACT通常是包括符合以下条件之一的FDA 监管药物、生物或器械产品的介入研究:(1)该试验在美国有一个或多个地点;(2)该试验根据FDA研究性新药申请或研究性器械豁免进行;(3)试验涉及在美国领土制造并出口的用于研究的药物、生物制品或器械产品。14
第二,在规定的时间内启动的临床试验。符合“适用临床试验”(ACT)定义且在2007年9月27日之后启动,或在该日期或之前启动但截至2007年12月26日仍在进行中的研究需要注册。15
以下类型的研究不受PHS 法案第 402(j) 节注册和结果提交要求的约束,包括(1)研究药品或生物制品的第一期试验,包括将研究药品用作研究工具来探索生物现象或疾病过程的研究;(2)与健康无关的确定器械产品可行性的小型临床试验或测试原型机16的临床试验,其中主要结果测量与可行性有关;(3)不包括药物、生物或器械产品的试验;(4)非介入性(观察性)临床研究。但以上研究可根据《最终规则》自愿提交注册和结果信息。责任方自愿提交不受强制约束的临床试验信息的,须遵守 PHS 法第 402(j) 条及其规定的特定要求。17
NIH主任可主动要求提交临床试验信息以保护公众健康。18PHS 法案第 402(j)(4)(B)(i) 节特别授权部长“通知要求”提交临床试验信息。19被要求提交的临床试验信息应当不迟于主任通知的日期之30天在ClinicalTrials.gov网站提交,除非责任方申请延迟结果信息提交证明。20
2.2. 责任方
《最终规则》规定必须有一个(且只有一个)责任方来提交有关ACT的信息。21责任方(responsible party)指临床试验的发起人(sponsor)22,或者临床试验的受让人(grantee)、承包商(contractor)以及被授权人(awardee)指定的主要研究者(principal investigator)。主要研究者应当是负责进行试验且有权访问和控制试验数据的研究人员,主要研究人员应当具有发表实验结果的权利,有能力满足《最终规则》对临床试验信息报送的全部要求。对于非临床试验的器械产品的儿科上市后监测,责任方是根据FDA命令对器械产品进行儿科上市后监测的实体。23
3. 临床试验数据注册与结果报告
《最终规则》旨在保障公众知情权,提高临床试验可重复性和安全性,改善为临床护理提供信息的证据基础,提高药品与医疗器械开发效率,改进临床研究时间并建立公众对临床研究的信任。为此《最终规则》详细规定了如何提交临床试验数据信息,以实现最终目的。
1) 试验注册和注册信息提交:
任何在2007年9月27日之后启动的或者部分特殊的2007年9月27之前启动至2007年12月26日仍在进行的ACT必须注册,并应当在第一位人类受试者注册后21日内提交临床试验注册信息。24其中2007年9月27日之前开始并在2007年12月26日仍在进行的涉及“严重或危及生命的疾病或病症”的试验必须在2007年12月26日之前提交注册信息,不涉及“严重或危及生命的疾病或病症”的试验需要在2008年9月27日之前提交注册信息。25ACT开始日期从第一个人类受试者招募之日起算,但非临床试验的器械产品进行儿科上市后监测开始日期为FDA批准实施监测计划之日。26
提交的临床试验注册信息至少每12个月更新一次,信息发生变更后应在30日内更新,对明显错误、缺陷或者不一致信息以及其他更正内容,责任方应当在收到电子通知之日起15日内更正临床试验注册信息,25日之内更正结果信息。27
NIH负责注册信息的公布。对于适用药物的临床试验,NIH主任将于提交信息之日起30日内在ClinicalTrials.gov网站上公开适用药物临床试验的临床试验注册信息(某些行政管理数据除外)。28而使用器械的临床试验则时间较晚。对于已经批准或经认可的器械产品的临床试验,NIH主任在可行的情况下应尽快在网站公布注册信息,尽快的解释应不晚于发布临床结果信息后的30个工作日。29未被批准或认可的器械产品的信息,公布时间将延迟到FDA批准或认可该器械产品之日起30日内。2017年1月18日之后启动的ACT,如果是未经批准和认可的器械产品,可以根据11.28(a)(2)(i)(Q)规定,授权NIH主任公布其临床试验注册信息,否则需待至产品经批准。30
2) 结果信息提交
i. 适用范围
研究完成后,ACT责任方必须提交该临床试验的结果信息,除非经批准豁免。研究完成被定义为“研究已正常结束,参与者不再接受检查或治疗;或者研究已被终止,不会恢复,参与者不再接受检查或治疗,参与者最后一次访问网站31。研究完成后,研究人员将清理数据,形成用于创建完整可分析数据的数据集,执行预先指定的附加分析。
所有完成日期在2007年12月26日之后的已批准、许可或清除产品(cleared products)的ACT都需要提交结果信息。《最终规则》还将结果信息提交的要求扩展到未经FDA批准和许可的药品(包括生物制品)和器械产品的ACT。对于主要完成日期在2017年1月18日或之后且研究产品未经FDA批准、许可,需要按照42CFR第11部分的规定提交结果信息。而主要完成日期在2017年1月18 日之前的未经批准的ACT,责任方当在不迟于产品获得批准后的30天年内提交结果信息。32
ii. 提交时间
一般而言,临床试验结果信息必须在临床试验主要完成日期后1年内提交。如果寻求新用途的批准和许可,则可以申请延迟提交结果信息,但不迟于2年。如果主要完成日期之前尚未收集到次要结果测量或其他不良事件信息,责任方必须在最终收集结果日期一年后提交结果信息,无论其方案是否终止。33
iii. 结果信息公开
除了根据PHS法案第402(j)(4)(A) 节和 § 11.60 提交的临床试验结果信息外,主任将在不迟于提交日期后 30 个日历日内在ClinicalTrials.gov上公开临床试验结果信息提交。
iv. 豁免提交结果信息
主要完成日期为2017年1月18日之后的ACT的责任方可以在提交结果信息截止日期之前向NIH机构提交就某一部分信息数据的豁免请求。如豁免请求被拒绝,申请方可在受到拒绝通知之日起30日内向NIH提交上诉。如该豁免申请被批准,NIH主任将在临床试验记录中注明本部分要求的特定要素已被豁免,部长将书面通知国会的相关委员会,并在不迟于授予任何豁免后的30个日历日内解释授予豁免的原因。34
3) 不合规的法律后果
i. 民事或刑事司法责任
未能遵守根据《公共卫生服务法》第 402(j) 节要求将违反《联邦食品、药品和化妆品法》规定。《联邦食品、药品和化妆品法》第331(jj)规定未提交402(j)(5)(B)规定的伴随药品、生物制品和器械提交的认证或者提交虚假证明35,未提交第402(j)条规定的临床试验信息或提交虚假或误导性临床试验信息,应承担民事或刑事责任。36
ii. 民事罚款
任何违反《联邦食品、药品和化妆品法》301(jj)节规定的将被处以一次诉讼中裁定的违法行为应受到不超过10000美元的民事罚款。发出通知后30天内为纠正的,除罚款外,还将每天被处以不超过10000美元的罚款,直到违规行为纠正。37
iii. 联邦拨款资助活动
根据《公共卫生服务法》第402(j)(5)(A)节规定,任何由健康和公众(Department of Health and Human Services)服务部资助的ACT,其拨款申请与研究进度报告表中应当包含责任方所提交注册和结果的证明。如果责任方未提交临床试验注册和结果信息,则不可发放资助款项,受资助者应当在30天内纠正不符合规定的情况并提交所需的临床试验信息。38
3
NIH《关于传播NIH资助的临床试验信息的最终政策》
为最大限度地发挥临床试验的价值,负责任的传播来自NIH资助的临床试验的信息,促进医疗行业进步,美国国立卫生研究院(NIH)出台了与《最终规则》配套的临床试验数据共享政策,即《关于传播NIH资助的临床试验信息的最终政策》(NIH Policy on the Dissemination of NIH-Funded Clinical Trial Information,以下简称《最终政策》)。该政策是《最终规则》的补充,所有受NIH资助的研究人员应当在遵守《最终规则》的同时遵守《最终政策》。
相较于《最终规则》,《最终政策》扩大了向ClinicalTrials.gov提交的信息适用范围。《最终规则》适用于受FDA监管的药物、生物和器械产品的介入研究,未包括未受监管的的临床研究,如药物和生物制品的一期试验、器械产品的小型可行性研究等,但其仍对医学科学进步、减少重复实验、提升临床试验透明度具有重要作用。NIH《最终政策》则针对所有受其资助的临床试验研究团队,无论其研究阶段、干预类型、是否受FDA法规约束,都应当向ClinicalTrials.gov注册并提交相关结果信息数据。但该政策不适用于仅使用NIH支持的基础设施但未获得NIH资金支持其行为的临床试验。39
《最终政策》细化了《最终规则》提交数据与责任承担规制。《最终政策》要求向NIH申请资助的申请人应当提交一份《传播受NIH资助的临床试验信息计划》并在研究过程中遵守所有条件。根据临床试验是否属于ACT,受资助者和研究人员应当承担以下责任,并由NIH将在ClinicalTrials.gov上公开发布注册信息和结果信息:
- 如果NIH资助的临床试验是法规规定的ACT,且受资助者或研究者为责任方,受资助方或研究者将确保满足所有监管要求。
- 如果NIH资助的临床试验是法规下ACT,但受资助者或研究者不是责任方,受资助方或研究者将与责任方协调,以确保满足所有监管要求。
- 如果NIH资助的临床试验不是法规规定的ACT,受资助者或研究者将负责执行任务并满足法规规定的时间表。
不遵守《最终政策》后果更为严重。遵守《最终政策》的要求作为NIH资助条件的一部分应当被严格遵守,如不符合《最终政策》规定,将NIH可能采取终止研究、标记不合格、阻止提交绩效进展报告40或强制执行等措施。如符合《联邦法规法典》第45章第75.371节违规,可能由HHS采取暂停资助或更为严厉的措施。41若NIH资助的临床试验落入FDA监管的ACT将导致上文《最终规则》民事、刑事以及资助活动的执法措施。42

此外,出于对《最终政策》的补充和完善,确保最大限度地提高数据管理和研究透明度,NIH不断增加新的对临床试验注册和结果信息提交的政策。如2017年NIH颁布了《将妇女和少数民族纳入临床研究的 NIH 政策和指南》(NIH Policy and Guidelines on the Inclusion of Women and Minorities as Subjects in Clinical Research)要求在 Clincialtrials.gov 注册时,适用的 NIH 定义的 III 期临床试验必须根据先前证据的要求以及NIH 关于纳入妇女和少数民族的政策和指南中的解释,具体说明性别和种族/民族的结果作为临床研究的对象。对于适用的 NIH 定义的 III 期临床试验,向 Clinicaltrials.gov 提交的结果必须包括根据先前证据的要求按性别/性别和种族/民族进行的有效分析结果。43NIH将持续支持临床试验数据共享,最大程度的利用现有数据,造福公众健康。
4 临床试验数据共享评价
临床试验注册和结果信息提交制度强制要求符合特定条件的临床试验研究团队公布其在研究数据和结果信息,并通过严格审查机制保障数据研究透明度和数据质量,推动了医疗健康数据共享,是国际开放科学的重要组成部分,其意义深远。
1. 积极影响
临床试验注册制度能够有效提升检索信息的便利程度,从而保障公众知情权。ClinicalTrials.gov创建了一个单一的、集中的数据库来识别研究任何疾病的干预措施的试验,其高度结构化的数据以及搜索引擎,使公众能够通过各种方式搜索其需要的试验信息,了解研究的效用及相关产品。网站注册的每一研究机构均具有唯一的ID标识及名称链接,用户可以检索到该机构临床试验生命周期的所有相关数据信息,即使不同临床试验企业在不同的开发周期使用不同的名称,无论其产品是否经批准上市,公众都能够充分了解临床试验方案和预计结果。因此共享数据的过程清晰、透明和负责,保障了公众知情权,也进一步提高公众信任。
临床试验数据共享促进科学研究与公共健康研究。科学进步与完整、客观、不偏倚的研究报告密切相关。临床试验注册制度完善了数据库共享数据规则,相较于单一出版物公开,能有效提高公开数据的完整性和公正性。调查显示出版物更倾向于公开积极效果,隐匿部分数据和研究结果,从而导致偏倚的结论甚至过于夸大效应量。44临床试验注册和结果信息提交制度则要求汇总提交临床试验生命周期产生的多类数据,包括临床试验方案、受试者招募和注册、原始数据和计算机处理的可分析数据集等能有效减少数据偏倚,为二次分析提供客观的数据资料。
临床试验数据注册和结果信息提交制度有效保障受试者人权。需要提交的临床试验注册信息包括研究类型、干预类型、人类受试者保护审查委员会状态、人类受试者招募信息、受试者知情同意等文件,并要求第一个人类受试者注册之日起及时提交注册信息,临床试验过程中创建试验及其结果的公共记录来确保他们的参与受到尊重。知情同意规则是临床试验数据共享制度保护人类受试者的核心措施。对受试者进行干预实验应当遵循《贝尔蒙特报告》(The Belmont Report)规定的伦理道德原则,尊重人类受试者自主权,尽最大程度减少威胁或者不当影响,以适当的方式使受试者了解研究重要信息。45同时临床试验必须符合《赫尔辛基宣言》(Declaration of Helsinki)要求,促进试验注册和结果传播同时采取一切预防措施保护研究对象的隐私及其个人信息的机密性。46
2. 隐私风险
ClinicalTrials.gov作为公共网站,允许任何人发布临床试验数据,也允许任何人通过访问网站检索相关数据信息。而临床试验数据以人类受试者数据为核心,提交的注册信息和结果信息中包含大量未去识别化的生物样本和私人信息47,虽然临床试验单位(The clinical trials enterprise)48应遵守《人类受试者保护联邦政策》(Federal Policy for the Protection of Human Subjects (‘Common Rule’)),遵守知情同意规则,但可识别信息的共享和二次分析将对人类受试者隐私和尊严产生极大威胁。如精神类疾病、遗传性疾病等研究数据将产生不可预见的超出合理范围的歧视或者社会风险。
参与者注册是临床试验核心数据获得的起点,期间获得的原始数据和可分析数据集均可能产生隐私风险。临床试验数据来自参与研究的患者和健康志愿者。原始数据是在第一个参与者注册和研究完成之间收集的。在试验过程中,原始数据被提取、编码和转录,参与者活动结束后,数据将整理到可分析数据集中。个人参与者原始数据是研究人员对个体参与者进行干预试验的直接或抽象观察结果。共享原始数据相比经处理的数据,其最能反映研究观察结果,可有效减少数据处理错误、缺陷或者误差所导致的数据质量下降问题,也可减少数据欺诈、数据偏见等现象。然而,原始数据庞大而复杂,其中包含参与者病史、基因组序列、成像结果或自我报告等涉及个人隐私与尊严的敏感数据,共享原始数据具有极大隐私风险。
完整的可分析数据集并不是已经全部处理和分析过的数据的集合,相反,其数据很大一部分未被使用,因而称为可分析数据集,是试验中最有用的数据集。可分析数据集共享可使其他研究者及公众获得未经分析和发表的数据,并允许其他研究人员重现原始分析,对主要研究目标进行替代的、科学有效的分析。此外,共享可分析数据允许通过额外的二次分析进行进一步的科学发现,以及对其他研究的一般假设进行探索性研究,对社会和医学研究产生巨大推动作用。然而,可分析数据集的共享虽非原始数据,但其共享仍具有一定隐私风险。数据集中的信息越详细,便越容易识别该数据集中的个体,重新识别的风险也在增加。
因而,随着大数据、算法等技术的发展,仅知情同意制度并不能全面规避隐私风险,美国各机构仍需要制定更合理、更完善的人类受试者隐私保护制度。
5 总结
美国临床试验注册和结果信息提交制度以ClinicalTrials.gov网站为核心,除《2007年食品和药品管理局修正法案》《临床试验注册及结果信息提交最终规则》《人类受试者保护政策》等法律法规外,NIH、退伍军人事务部(Department of Veterans Affairs)49、以患者为中心的结果研究所(Patient-Centered Outcomes Research Institute (PCORI)50)等机构和部门亦发布相关政策,同时国际医学期刊编辑委员会(The International Committee of Medical Journal Editors(ICMJE))51、世界卫生组织(World Health Organization)52提出临床试验数据注册和信息提交要求,共同促进负责任的临床试验数据公开,进一步提升了美国临床试验领域公信力。
临床试验产品及治疗方法的上市需要合理的临床试验方案以及全球注册策略,美国依据ClinicalTrials.gov网站建立了相对统一的数据共享注册制度,也对进入美国的临床试验产品提出了新要求53,扩大了国际影响力。这种数据共享制度和规则值得我们进一步研究。
参考文献:
[1] COVID-19 Clinical Trials, https://covid19.nih.gov/clinical-trials.
[2] FDAMA(1997) SEC. 113.(a)(2),目前被编入42 U.S.C. 282(i), PHS Act 402(i)).
[3] 42 USC 282(j).
[4] Richard Tadd, 美国临床试验数据库(ClinicalTrials)注册流程与填写要求, https://www.linkedin.com/pulse/%E7%BE%8E%E5%9B%BD%E4%B8%B4%E5%BA%8A%E8%AF%95%E9%AA%8C%E6%95%B0%E6%8D%AE%E5%BA%93clinicaltrials%E6%B3%A8%E5%86%8C%E6%B5%81%E7%A8%8B%E4%B8%8E%E5%A1%AB%E5%86%99%E8%A6%81%E6%B1%82-richard-tadd.
[5] 如2013年欧盟药品管理局(EMA)发布了新版本的欧洲临床试验数据库(欧盟医药临床试验法规自今年1月31日起实施 https://www.qservegroup.com/ch/zh/i1126/clinical-trials-regulation–ctr-for-medicinal-products-in-application-on-31-january-2022 )。
[6] National Institutes of Health’s “ClinicalTrials.gov” Web Site Wins Prestigious Award, https://www.nlm.nih.gov/archive/20120510/news/press_releases/clintrialsaward04.html.
[7] FDAAA 801 and the Final Rule https://clinicaltrials.gov/ct2/manage-recs/fdaaa.
[8] 42 CFR 11.10.
[9] 21 U.S.C. 360(k), 21 U.S.C. 360e, 21 U.S.C. 360j(m).
[10] 21 U.S.C. 3601.
[11] 21 CFR part 3 https://www.ecfr.gov/current/title-21/chapter-I/subchapter-A/part-3.
[12] 21 U.S.C. 355.
[13] 42 U.S.C. 262.
[14] Checklist for Evaluating Whether a Clinical Trial or Study is an Applicable Clinical Trial (ACT) https://prsinfo.clinicaltrials.gov/ACT_Checklist.pdf.
[15] 42 CFR 11.22.
[16] 原型机是指尚未准备好大量生产和销售的新的机器或者器械、设备未准备。A prototype is a new type of machine or device which is not yet ready to be made in large numbers and sold.(https://www.collinsdictionary.com/dictionary/english/prototype-device)
[17] 42 CFR 11.60.
[18] 42 CFR 11.62.
[19] A Notice by the National Institutes of Health on 04/30/2009 https://www.federalregister.gov/documents/2009/04/30/E9-9699/statement-of-delegation-of-authority.
[20] 42 CFR 11.62(c)(1).
[21] 42 CFR 11.4(c).
[22] 21 CFR 50.3.
[23] 42 CFR 11.10(a).
[24] 42 CFR 11.24(a).
[25] 42 CFR 11.24(b).
[26] Clinical Trials Registration and Results Information Submission, https://www.federalregister.gov/d/2016-22129/p-1299.
[27] 42 CFR 11.64.
[28] 42 CFR 11.35(a).
[29] 42 CFR 11.35(b)(1).
[30] 42 CFR 11.35(b)(2).
[31] European Union Clinical Trials Register, 2011.
[32] Which Trials Must Have Results Information Submitted to ClinicalTrials.gov?, https://clinicaltrials.gov/ct2/manage-recs/fdaaa.
[33] 42 CFR 11.44.
[34] 42 USC 282(j)(3)(H)、42 CFR 11.54.
[35] 21 USC 331(jj)(1) .
[36] 21 USC 331(jj)(2)(3).
[37] 21 USC 333(f) )(3).
[38] 42 U.S.C. 282(j)(5)(A).
[39] NIH Policy on Dissemination of NIH-Funded Clinical Trial Information, https://grants.nih.gov/grants/guide/notice-files/NOT-OD-16-149.html.
[40] Guidance electronic Research Administration (eRA) Research Performance Progress Report (RPPR) Submission Validations for Clinical Trial Registration and Results Reporting, https://grants.nih.gov/grants/guide/notice-files/NOT-OD-22-008.html.
[41] 45 CFR 75.371.
[42] 42 USC 282(j), 42 CFR Part 11.
[43] NIH Policy and Guidelines on the Inclusion of Women and Minorities as Subjects in Clinical Research, https://grants.nih.gov/grants/guide/notice-files/NOT-OD-18-014.html.
[44] Ross JS, Tse T, Zarin DA, Xu H, Zhou L, Krumholz HM. Publication of NIH funded trials registered in ClinicalTrials.gov: cross sectional analysis. BMJ. 2012 Jan 3;344:d7292. doi: 10.1136/bmj.d7292. PMID: 22214755; PMCID: PMC3623605.
[45] The Belmont Report, https://www.hhs.gov/ohrp/regulations-and-policy/belmont-report/read-the-belmont-report/index.html.
[46] WMA DECLARATION OF HELSINKI – ETHICAL PRINCIPLES FOR MEDICAL RESEARCH INVOLVING HUMAN SUBJECTS, https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/.
[47] 《人类受试者保护联邦政策》(A子规则:保护人类研究对象的基本 HHS 政策)第102(e)条规定:私人信息包括关于在个人可以合理预期不会发生观察或记录的情况下发生的行为的信息,以及个人为特定目的提供的信息,并且个人可以合理预期将不得公开(例如,医疗记录)。可识别隐私信息,是指调查者已经或者可能容易识别主体身份或者与信息相关联的隐私信息。可识别的生物样本是指受试者的身份已经或可能很容易被研究者确定或与生物样本相关联的生物样本。(45 CFR 46.102)
[48] 临床试验企业涵盖所有临床试验及其应用。它包括参与研究的流程、机构和个人,以及最终将临床试验结果应用于护理环境的人员。National Academies of Sciences, Engineering, and Medicine. 2015. Sharing Clinical Trial Data: Maximizing Benefits, Minimizing Risk. Washington, DC: The National Academies Press. https://doi.org/10.17226/18998.
[49] Department of Veterans Affairs (VA) Clinical Trial Registration and Results Submission Policy, https://www.research.va.gov/resources/ORD_Admin/clinical_trials/.
[50] Patient-Centered Outcomes Research Institute (PCORI) Process for Peer Review and Public Release of Results, https://www.pcori.org/news-release/pcori-board-adopts-process-peer-review-and-public-release-research-findings.
[51] Clinical Trials, https://www.icmje.org/recommendations/browse/publishing-and-editorial-issues/clinical-trial-registration.html.
[52] 国际临床试验注册平台,https://www.who.int/zh/publications/m/item/international-clinical-trials-registry-platform。
[53] 国产新药赴美上市审评,FDA的“公开课”讲了什么?,https://finance.sina.cn/2022-02-23/detail-imcwipih4994772.d.html。
 关注微信公众号
关注微信公众号
